Introduction
Serum calcium (Ca2+) value aberrations represent a group of disorders that cannot be seen as isolated biochemical events; rather, they encompass failures of the integrated endocrine system. Serum calcium values can often be misinterpreted if deciphered in isolation, as they may take the disguise of normalcy even when the mineral repository and regulatory axis is under profound debilitation, and the fact that they can elicit a unique capacity to appear abnormal while the underlying disease remains invisible. The study of metabolic bone disorders treats calcium not as a diagnosis but as a signal demanding source attribution.
The Inextricable Triad
The homeostasis of calcium, phosphate, and parathyroid hormone is set through mutual feedback mechanisms, and the values of all three elements have little diagnostic value when considered independently. PTH uses calcium as a cue, when calcium falls, PTH rises, mobilizing skeletal calcium, stimulating renal 1-alpha hydroxylase, and suppressing phosphate reabsorption. The typical pattern of primary hyperparathyroidism, i.e., hypercalcemia, hypophosphatemia, and an elevated or inappropriately normal PTH, makes diagnostic sense only when understood as a failure of the CaSR-mediated feedback set point rather than as a parathyroid gland producing PTH in error. A receptor expressed abundantly on the parathyroid gland and renal tubular cells, belonging to the family of C GPCR, which responds to calcium levels; rising calcium levels lead to suppression of PTH secretion, but in primary hyperparathyroidism, this suppression fails. The laboratory profile that follows is a physiologic footprint, not the disease itself.
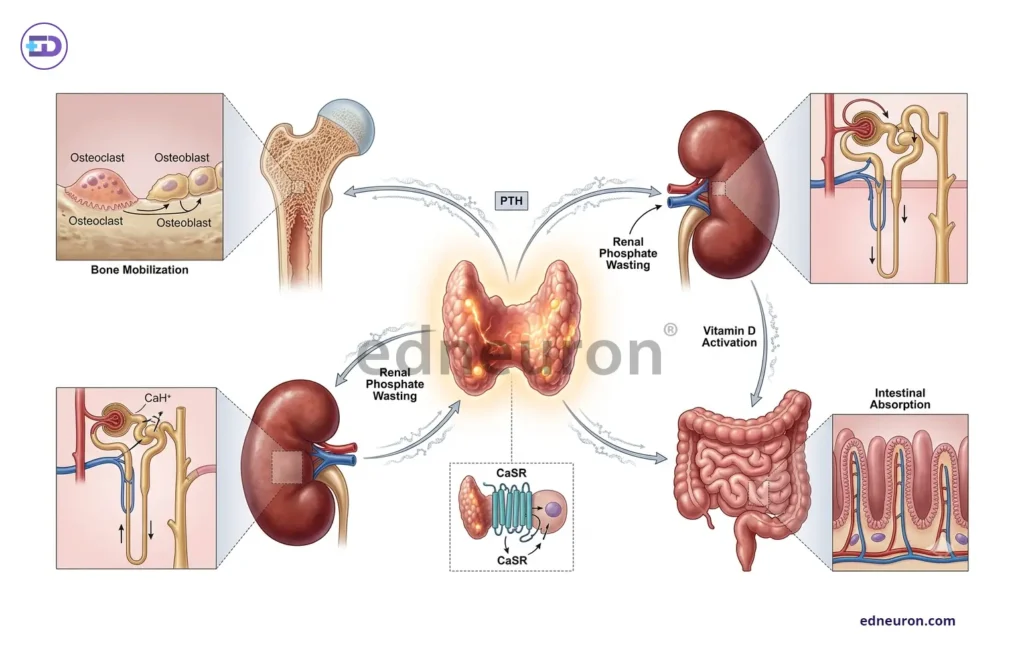
Fig 1 : The Calcium–Phosphate–PTH Regulatory Axis.
Vitamin D and the Masking Problem
Vitamin D deficiency represents a major diagnostic confounder for clinicians in cases with underlying primary hyperparathyroidism. The physiological action of vitamin D is to increase intestinal absorption. of calcium, but in patients with underlying primary hyperparathyroidism, the calcium-lowering effect of vitamin D deficiency by suppressing intestinal calcium absorption, thereby reducing the calcium available to stimulate CaSR-mediated inhibition of PTH secretion, can offset the expected hypercalcemia. Concomitant vitamin D deficiency thus normalizes serum calcium in patients with underlying primary hyperparathyroidism, creating an apparent normocalcemic hyperparathyroidism or masking established disease entirely. In this setting, when we correct the Vit. D, it leads to a return of the typical biochemical presentation and frequently unmasks the true diagnosis. This is a predictable consequence of all: vitamin D is not simply a calcium-raising hormone but rather a modulator of the gain-of-function of the entire PTH-calcium feedback loop. Assessing 25-hydroxy vitamin D before interpreting elevated PTH is not just optional in any rigorous evaluation, but it is the step without which the PTH result cannot be rendered interpretable.
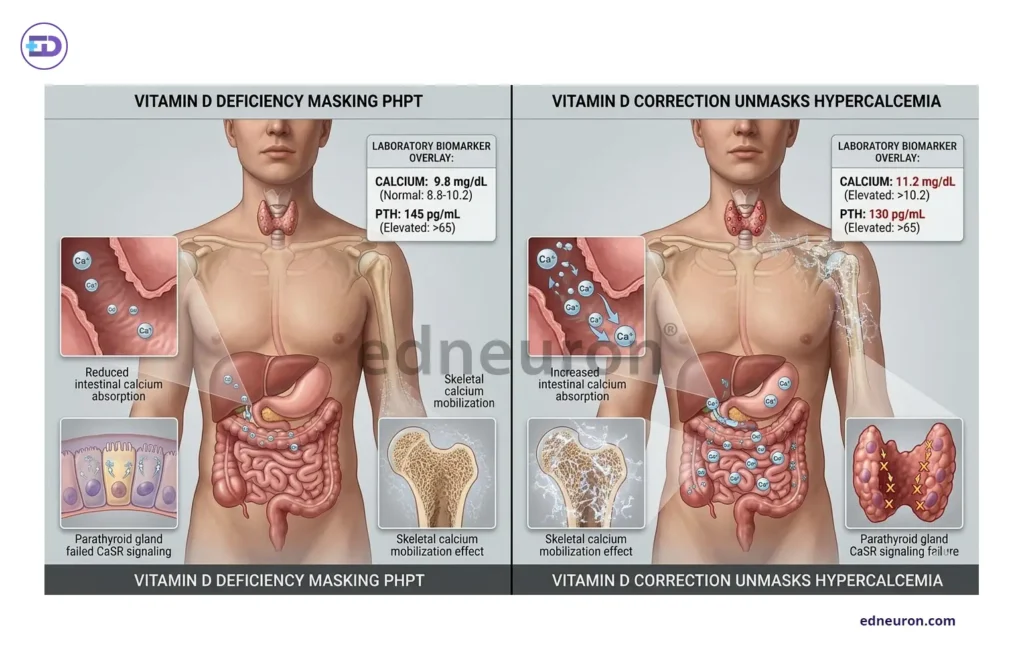
Fig 2 : Vitamin D and the masking of primary hyperparathyroidism.
FGF23, Phosphate, and the Renal Dimension
The discovery and eminent characterization of fibroblast growth factor 23 broadened our understanding of the calcium regulatory axis into a new network with phosphate at its core, going beyond the classical dyad of PTH-vitamin D and with profound clinical implications.FGF23, produced by osteocytes in response to phosphate load and the active form of vitamin D, i.e., 1,25-dihydroxcholecalciferol, acts on the renal tubules in concert with its obligate co-receptor alpha-Klotho to suppress phosphate reabsorption and inhibit 1-alpha hydroxylase, creating a counter-regulatory loop that is not explained by PTH alone. In chronic kidney disease, in response to phosphate retention, an adaptation characterized by rising FGF23 and falling Klotho levels may signify an early adaptation that precedes clinical hyperphosphatemia by years; the consequent suppression of calcitriol production drives the development of secondary hyperparathyroidism through a functionally distinct pathway from simple calcium deficiency. The current KDIGO CKD-MBD recognizes two major clinically dominant disease phenotypes: one characterized by fracture risk and the other by vascular calcification. Both of these require an individualized interpretation of the same biochemical panel. Renal function needs to be thoroughly assessed, as applying standard thresholds without due diligence constitutes a structural diagnostic error rather than a clinical judgment call.
Bone turnover markers, such as serum P1NP (the formation index) and CTX (the resorption index), add the temporal dimension that static calcium values cannot provide. The bone does not represent a static organ; rather, its architecture is ever so dynamic, and these markers provide a biochemical insight into bone turnover dynamics and distinguish between a state of accelerated uncoupled remodeling, a feature of Paget’s disease and high-turnover osteoporosis, and a state of suppressed equilibrium, as in adynamic bone disease complicating advanced CKD or prolonged bisphosphonate use. Even with polar-opposite bone biology, the reflected calcium may be identical. Tumor-induced osteomalacia is driven by FGF 23 overproduction from a small phosphaturic mesenchymal tumor that may remain occult for years. Excess FGF23 suppresses phosphate absorption via PCT and inhibits renal 1-alpha-hydroxylase activity, which allows lacunae that promote renal phosphate wasting, hypophosphatemia, and inappropriately low calcitriol levels. Although the tumor itself is benign, it represents a rare but diagnostically elusive entity that illustrates the stakes of incomplete evaluation. The pivotal diagnostic clue is the phosphate levels, which are more often than not ignored. It presents with disabling bone pain and fractures, carries a 95% initial misdiagnosis rate, and these symptoms are attributed to more common conditions such as osteoporosis, fibromyalgia, inflammatory disease, vitamin D deficiency, degenerative spine disease, or even functional disorders, and resolves entirely with tumor resection, but only after the clinician thought to measure serum phosphate.
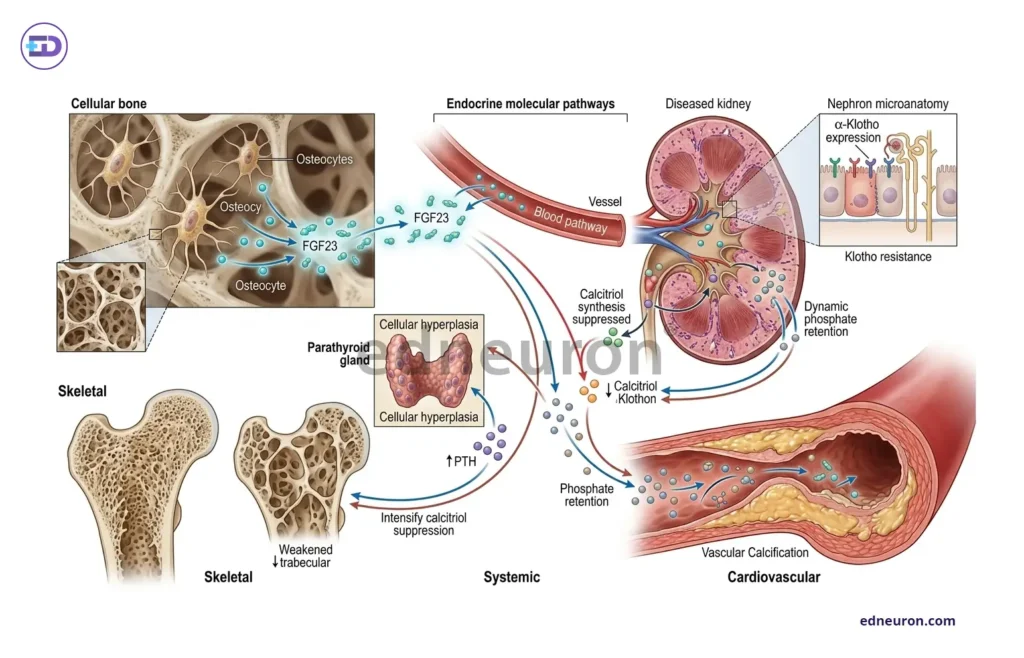
Fig 3 : FGF23-KLOTHO AXIS in CKD-MBD.
Conclusion
An evaluation of calcium–bone dysbalance requires an orderly assessment of the regulatory axis in the order that physiology demands, rather than isolated laboratory thresholds of the various biomarkers.
- Calcium status confirmed with albumin-corrected or ionized measurement.
- Phosphate pattern to contextualize PTH direction+PTH appropriateness assessed against both calcium and vitamin D status.
- Renal function is to be integrated before any mineral threshold is applied.
- Bone turnover markers to characterize skeletal kinetics.
In clinical cases with hypomagnesia, calcium replacement may prove to be ineffective as hypomagnesia causes an impaired secretion of PTH, thereby in hypocalcemic presentations, magnesium status also needs to be evaluated. No single result reaches the diagnosis from which the others are absent. Metabolic bone disease is solved not by chasing calcium values, but by reconstructing the endocrine conversation from which those values emerged.
References
- Williams C, Anastasopoulou C, Sapra A. Biochemical Markers of Osteoporosis. Treasure Island (FL): StatPearls Publishing; 2026 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK559306/
- Endocrine Society. Clinical Practice Guideline: Bone Health and Osteoporosis. 2022. Available at: https://www.endocrine.org/clinical-practice-guidelines/bone-health-and-osteoporosis.
- Yuan M, Ma T, Fan Z, Li J, Zhang S. The calcium-sensing receptor: a comprehensive review on its role in calcium homeostasis and therapeutic implications. Am J Transl Res. 2025 Mar 15;17(3):2322-2338. PMID: 40226019.
- Yedla N, Kim H, Sharma A, Wang X. Vitamin D Deficiency and the Presentation of Primary Hyperparathyroidism: A Mini Review. Int J Endocrinol. 2023 Dec 11;2023:1169249. PMID: 38115826.
- Razzaque MS. The FGF23-Klotho axis: endocrine regulation of phosphate homeostasis. Nat Rev Endocrinol. 2009 Nov;5(11):611-9. PMID: 19844248.
- KDIGO. CKD-MBD Controversies Conference Final Report. Kidney International. 2025. Available at: https://kdigo.org/
- Salvatore Minisola, Seiji Fukumoto, Weibo Xia, Alessandro Corsi, Luciano Colangelo, Alfredo Scillitani, Jessica Pepe, Cristiana Cipriani, Rajesh V Thakker, Tumor-induced Osteomalacia: A Comprehensive Review, Endocrine Reviews, Volume 44, Issue 2, April 2023, Pages 323–353,
- Boyce BF, Xing L. Functions of RANKL/RANK/OPG in bone modeling and remodeling. Arch Biochem Biophys. 2008 May 15;473(2):139-46. PMID: 18395508.

Author: A P
A medical trainee with an emerging focus on translational and clinical research, with interests spanning surgical sciences, neuroscience, pediatrics, and immunology. Her academic trajectory reflects an effort to integrate molecular innovation with clinically relevant disease models, particularly in complex and high-burden conditions. Her research experience includes work in genome engineering, specifically in prime editing, exploring its therapeutic potential in precision medicine. She has also contributed to oncological research examining cholangiocarcinoma with brain metastasis, focusing on its clinical course and diagnostic challenges. In parallel, her work investigating stoma formation as an independent risk factor for acute kidney injury reflects an interest in perioperative and systemic complications. Academically, she has contributed to case-based and review-driven scholarship, including a case reports and interdisciplinary review articles. Her evolving interests in neurology, pediatrics, and immunology reflect a broader inclination toward understanding disease across systems—from molecular mechanisms to clinical outcomes—while maintaining a disciplined, evidence-based approach to patient care. MBBS (MS4) ABVIMS Dr. RML HOSPITAL New Delhi

