
Abstract
The discovery of APOL1 G1 and G2 risk variants, which account for a substantial proportion of the excess burden of focal segmental glomerulosclerosis and progressive kidney disease in individuals of West African ancestry, has produced a gigantic genetic tree of extraordinary power and a clinical implementation problem of equal complexity. APOL1 variants have the ability to identify disease risk and stratify patients, which the nominative labels like “hypertensive nephrosclerosis” cannot even approach. However, this precision is not all gold gilded land mine, incomplete penetrance, context-dependent second-hit biology, and the absence of widely available targeted therapy convolutes the moment of truth, pinpointing exactly which genotype becomes a clinically actionable diagnosis. This article argues that the central challenge of the discovery of APOL1 nephropathy is translational, demanding to know when genetic knowledge, unaccompanied by a proven therapeutic response, crosses the threshold of clinical obligation. The emerging data on inaxaplin suggest that the threshold may be approaching. Whether the field is ready for what follows is a separate and harder question.
A Genotype in Search of a Clinical Framework
When Genovese and colleagues published the association of APOL1 G1 and G2 risk variants with kidney disease in African Americans in 2010, the implications were immediately apparent and uncomfortable [FSGS odds ratio = 10.5 (95% CI 6.0–18.4); H-ESKD odds ratio = 7.3 (95% CI 5.6–9.5)]. APOL1 is a serum factor responsible for trypanosmal lysis. A genetic embodiment (Genetic variation at a locus in or near the MYH9 gene on chromosome 22) had been identified that explained the reason behind a major chunk of excess CKD burden that had long been attributed, often without biopsy confirmation, to hypertension and its renal end-stage kidney disease sequelae.
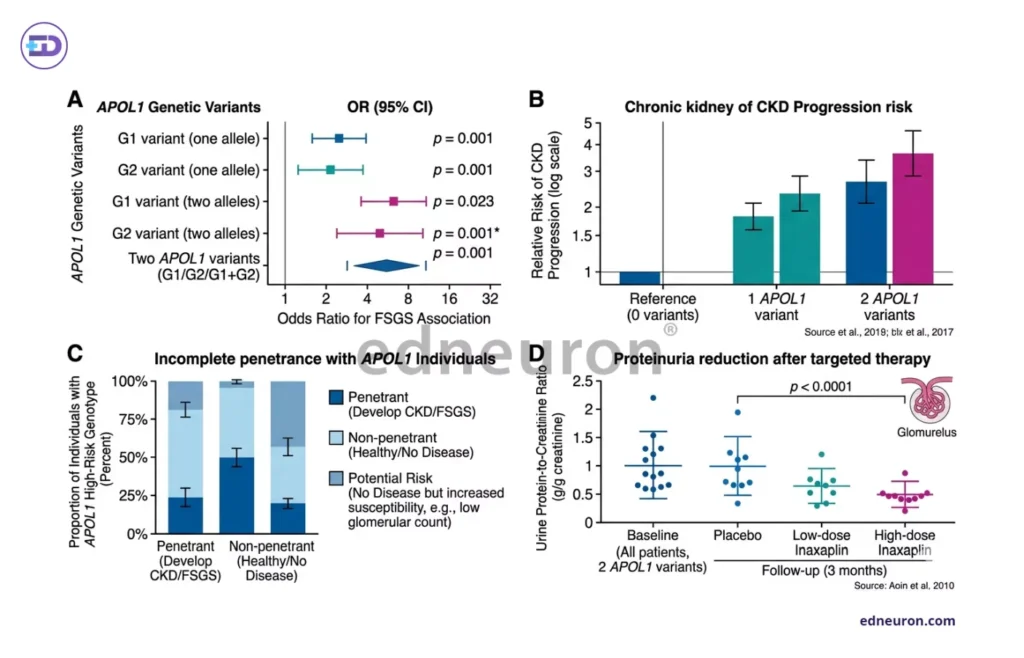
Fig 1 : A) The association between APOL1 risk alleles (G1 and G2) and susceptibility to kidney disease. B) CKD progression risk stratified by APOL1 genotype burden. C) Incomplete penetrance in APOL1-associated disease. D) Illustrative representation reduction in urine protein-to-creatinine ratio following selective APOL1 channel inhibition (e.g., inaxaplin).
The APOL1 finding did not merely add a risk factor to an existing diagnostic framework. It challenged the framework itself. ‘Hypertensive nephrosclerosis,’ that was exhaustively used as a label to describe any black patient with CKD presenting with a history of hypertension and virtually no other identifiable cause, in many cases described not a hemodynamic injury but a molecularly distinct podocytopathy in which elevated blood pressure is a cofactor rather than a cause. However, it must be kept in mind that not all arterionephrosclerosis patients of Afro-american ethinicity have APOL1 variation, and many patients of Asian and European descent exhibit FSG type nephropathy. But it is undeniable that the diagnostic language of nephrology, in this domain, has reflected historical assumptions more than conceptual precision.
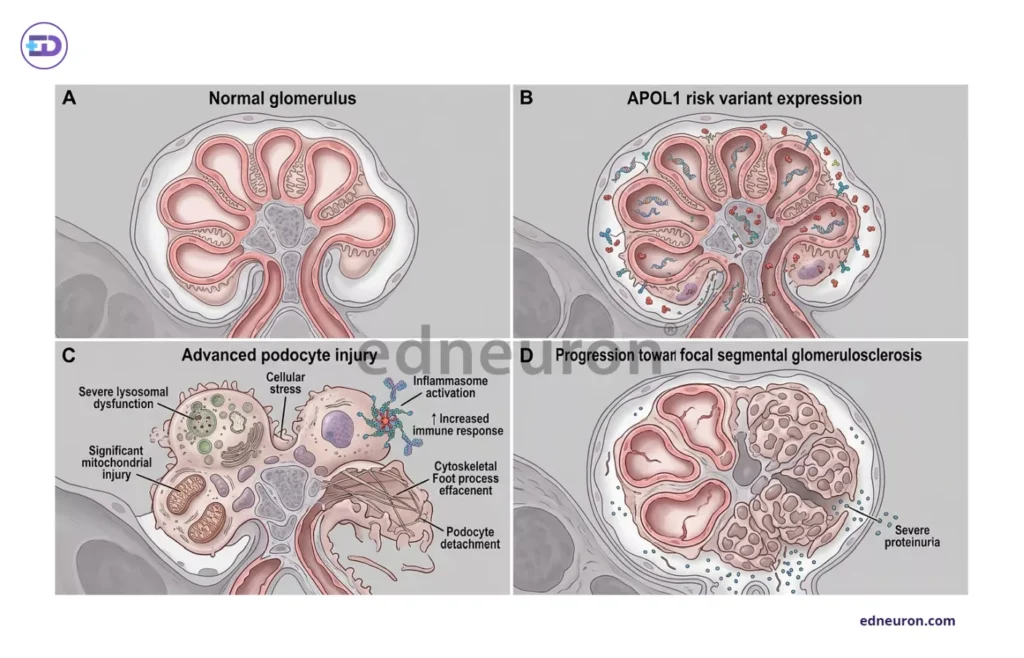
Fig 2 : Cellular Mechanisms of APOL1 Nephropathy..
Mechanism of the Disease
The question of whether APOL1 defines a disease entity or merely modifies susceptibility determines the scope of clinical obligation that a positive genotype creates. The current evidence bias is a distinct entity. APOL1 risk variants preferentially are responsible for causing focal segmental glomerulosclerosis and HIV-associated nephropathy, but are not associated with IgA or diabetic nephropathy, implying a different specific mechanism at work rather than generalized susceptibility. At the cellular level, the APOL1 risk-variant protein accumulates in podocytes, causing lysosomal disruption, mitochondrial function impairment, and NLRP inflammasome activation through pathways that are yet to be clearly discovered and properly delineated, oscillating between toxic gain-of-function and loss of protective biology.
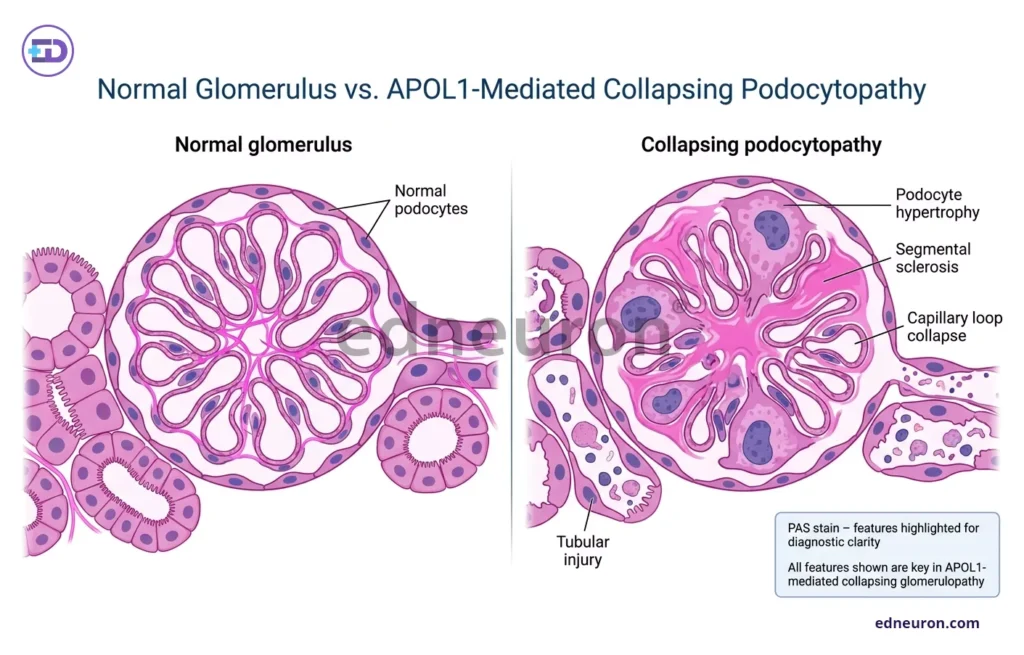
Fig 3 : Histological diagram of APOL1-mediated podocytopathy.
It was also found that such a high-risk genotype alone is insufficient, paving the way for another hypothesis-the second-hit hypothesis. Interferon signaling, viral infection, hemodynamic stress, or pharmacologic exposure activates APOL1 expression and precipitates podocyte collapse in a primed genotype. Such involvement of dynamic factors explains why penetrance is incomplete, and the same genotype can produce versatile disease phenotypes across individuals and decades. Such conditional variability makes clinical translation a very difficult task.
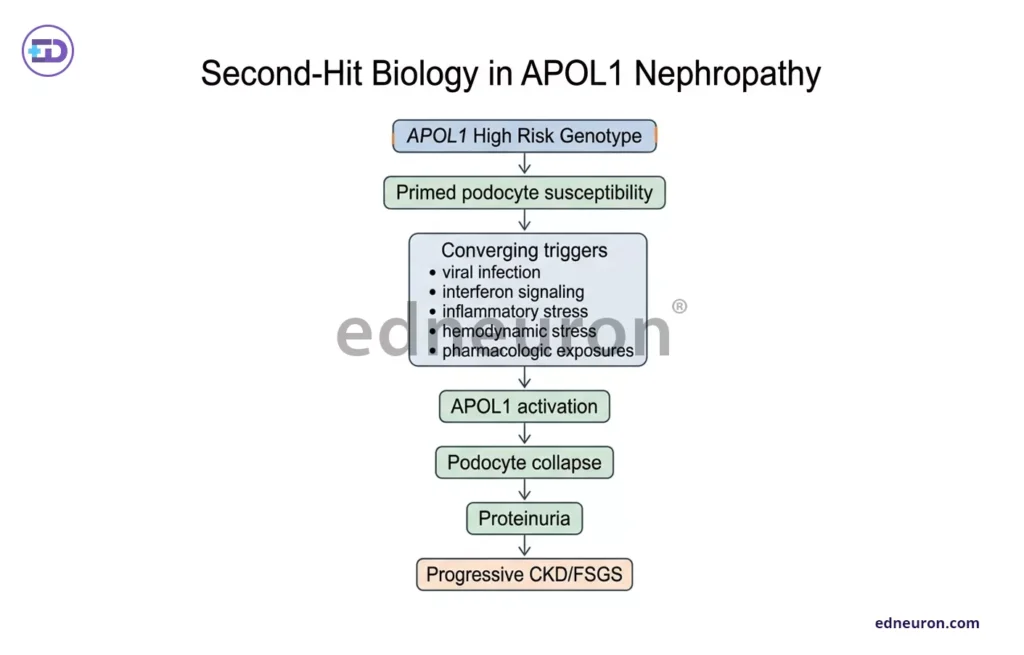
Fig 4 : The Second Hit Hypothesis.
The Precise Problem of Translation
Routine APOL1 genotyping in CKD evaluation is considered on 3 kaleidoscopic premises:
- The result can change diagnostic classification.
- It alters clinical management.
- Any harm of disclosure is proportionate to the informational benefit.
The first is now well-supported, with the reclassification of presumed hypertensive nephrosclerosis to APOL1-mediated glomerulosclerosis, important for the framework microcosm within which surveillance, biopsy thresholds, and donor evaluation decisions are made.
The second point is more so a static frostbite because the management of a high-risk APOL1 genotype without biopsy-confirmed active disease is not substantially different from standard optimization of blood pressure control, maximizing RAAS blockade, and heightened vigilance around second-hit exposures, including certain pharmacologic agents and intercurrent viral illnesses, the main motive being the strict amelioration of CKD symptoms.
The third introduces the fateful ethical requirements that precision genetics inevitably entails. Breaking the news of a genotype to the patient that predicts substantial lifetime risk without a proven disease-modifying response, yet that requires a well-established counseling infrastructure, cultural competence, and psychological support frameworks, is something nephrology has not systematically or institutionally built.
Inaxaplin and the Shifting Threshold
The clinical calculus of APOL1 genotyping is not static. Inaxaplin, a selective small-molecule inhibitor of APOL1 channel function, produced meaningful reductions in proteinuria in a phase 2 trial of patients with two APOL1 risk alleles and proteinuric kidney disease, providing the first pharmacological proof that APOL1 biology is therapeutically accessible. Such early safety data is reassuring, and if inaxaplin or a successor compound achieves success in GFR protection at scale, the threshold for genotyping will shift. This will enable the classifiers to then identify candidates for genotype-directed therapy, transforming APOL1 testing from a prognostic act into a therapeutic decision. That moment has not yet arrived, but the path is foreseeable, and the field awaits the phase 3 data on Inaxaplin. The obligation that emerges with clarity is the construction of a genotyping and counseling infrastructure to completion before that data lands.
Conclusion
APOL1 nephropathy is almost a musing in precision medicine, enquiring whether understanding the disease mechanism is itself a form of clinical action even before the therapeutic intervention develops. The reclassification of disease, the informed counseling of living kidney donors and in patients with LD (linkage disequilibrium), the avoidance of second-hit exposures, and the honesty of telling a patient what is actually damaging their kidneys rather than what is historically convenient to say are not trivial, invaluable gains in the absence of targeted therapy. They are the preliminary obligations that make targeted therapy meaningful when it arrives.
References
- Genovese G, Friedman DJ, Ross MD, et al. Association of trypanolytic ApoL1 variants with kidney disease in African Americans. Science. 2010 Aug 13;329(5993):841-5. PMID: 20647424.
- Kopp JB. Rethinking hypertensive kidney disease: arterionephrosclerosis as a genetic, metabolic, and inflammatory disorder. Curr Opin Nephrol Hypertens. 2013 May;22(3):266-72. PMID: 23470819.
- Bruggeman LA, O’Toole JF, Sedor JR. APOL1 polymorphisms and kidney disease: loss-of-function or gain-of-function? Am J Physiol Renal Physiol. 2019 Jan 1;316(1):F1-F8. PMID: 30332315.
- Giudicelli GC, De Souza CMB, Veronese FV, et al. Precision medicine implementation challenges for APOL1 testing in chronic kidney disease in admixed populations. Front Genet. 2022 Dec 15;13:1016341. PMID: 36588788.
- Smith JD, Agrawal A, Wicklund C, et al. Implementation of a culturally competent APOL1 genetic testing programme into living donor evaluation: A two-site, non-randomised, pre-post trial design. BMJ Open. 2023 May 15;13(5):e067657. PMID: 37188469.
- Egbuna O, Zimmerman B, Manos G, et al. VX19-147-101 Study Group. Inaxaplin for Proteinuric Kidney Disease in Persons with Two APOL1 Variants. N Engl J Med. 2023 Mar 16;388(11):969-979. PMID: 36920755.

Author: A P
A medical trainee with an emerging focus on translational and clinical research, with interests spanning surgical sciences, neuroscience, pediatrics, and immunology. Her academic trajectory reflects an effort to integrate molecular innovation with clinically relevant disease models, particularly in complex and high-burden conditions. Her research experience includes work in genome engineering, specifically in prime editing, exploring its therapeutic potential in precision medicine. She has also contributed to oncological research examining cholangiocarcinoma with brain metastasis, focusing on its clinical course and diagnostic challenges. In parallel, her work investigating stoma formation as an independent risk factor for acute kidney injury reflects an interest in perioperative and systemic complications. Academically, she has contributed to case-based and review-driven scholarship, including a case reports and interdisciplinary review articles. Her evolving interests in neurology, pediatrics, and immunology reflect a broader inclination toward understanding disease across systems—from molecular mechanisms to clinical outcomes—while maintaining a disciplined, evidence-based approach to patient care. MBBS (MS4) ABVIMS Dr. RML HOSPITAL New Delhi