
Abstract
AKI staging, based on urine output and creatinine levels, which has been the clinical standard for four decades, identifies kidney dysfunction, not kidney injury. Yet the transition from AKI to chronic kidney disease is governed not by how far creatinine rose, but by whether the injured tubular epithelium repaired adaptively or maladaptively. Patients who are positive for tubulointerstitial markers (KIM-1, NGAL, TIMP) and creatinine-negative, ‘subclinical’ AKI act as an accelerant to CKD; contradistinction to this are patients with high-stage creatinine AKI who achieve complete structural repair and may restore nephron integrity without long-term functional loss. This discussion wades through the tunnel of the AKI-to-CKD disease progression, which is fundamentally governed by epithelial cell-cycle arrest G2/M, loss of capillary density, tubular senescence, and self-sabotaging fibrogenic paracrine signaling, and finally proposes to reclassify AKI around structural injury biology and repair trajectories, which represents the necessary next step in precision nephrology.
When the Creatinine level Stages Kidney Dysfunction, Not Injury
The KDIGO defines a creatinine-based AKI staging system to create a reproducible, globally applicable, real-time signal of acute kidney dysfunction across heterogeneous clinical environments.
Reference- KDIGO Clinical Practice Guideline for Acute Kidney Injury. Kidney Int Suppl. 2012;2(1):1–138.

Table 1 : Criteria for acute kidney injury for children and adults (any one or more of the following within the corresponding time period).
Table 2 : Severity staging system for AKI.
It achieved its purpose of setting a global standard but utterly fails to comment on structural tubular injury, the characterization of repair quality, or the prediction of long-term renal fate. Creatinine is a glomerular filtration marker that rises when the loss of functional nephron mass exceeds what is required to maintain filtration. Being exquisitely sensitive to hemodynamic AKI, it shows a transient decline with afferent arteriolar constriction without tubular injury of any consequence. What creatinine lacks as a biomarker is sensitivity to structural tubular damage that occurs below the threshold of renal functional reserve, and it is functionally blind to the biology that determines whether injured tubular cells will regenerate, senesce, or ensue a vicious cycle of maladaptive repair, inflammation, and fibrosis. The concept of subclinical AKI makes such a blind spot precise. Newer, more sensitive biomarkers (elevated KIM-1, NGAL, or TIMP-2/IGFBP7) open a new window of risk: patients who are biomarker-positive and yet are creatinine-negative, the rate of translation from subsequent AKI to CKD is substantially higher.
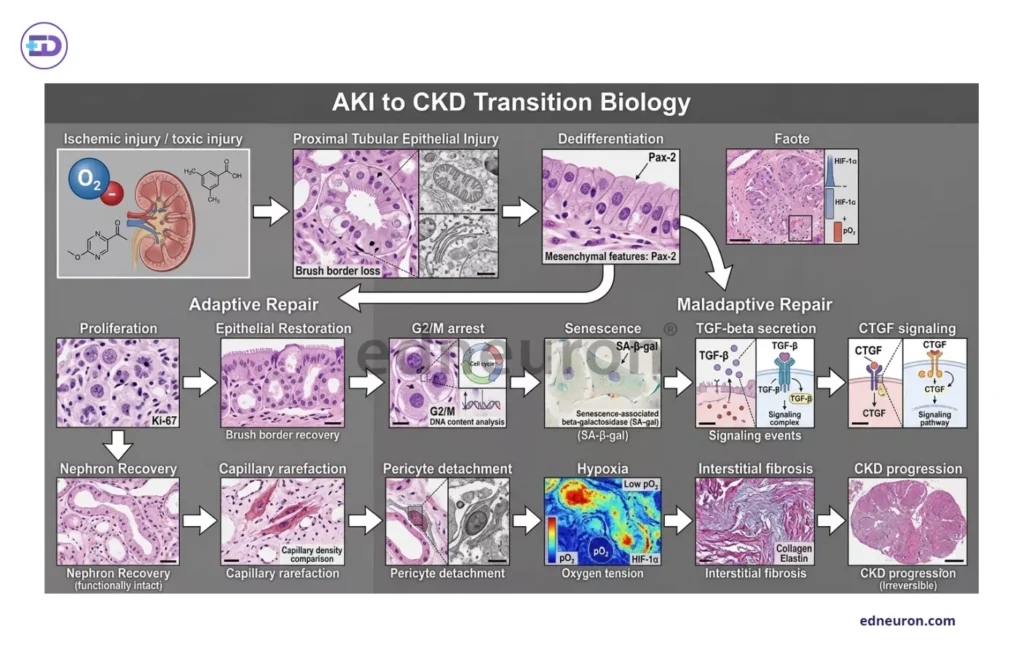
Fig 1 : Framework illustrating the biologic determinants of AKI-to-CKD transition.
Destiny of Repair Biology
The intellectual center and the key concepts of the AKI-to-CKD transition debate are not the magnitude of the initial injury but the fidelity of subsequent repair. Injured tubular epithelial cells have to redeem from a binary fate: either adaptive regeneration, in which dedifferentiated cells re-enter the cell cycle, proliferate, and restore functional epithelial integrity and the functional compensatory increase in GFR via hyperfilteration and glomerular hypertrophy; or maladaptive arrest, in which cells stall in G2/M, acquire a senescence-associated secretory phenotype, and begin constitutively secreting TGF-β, CTGF, and other profibrotic mediators that activate interstitial myofibroblasts. Such is the story of the predominance of tubulointerstitial fibrosis over glomerulosclerosis. The arrested epithelial population is a paracrine fibrosis factory, docking persistent DNA damage response signaling and uncoupled from the recuperative programs that would otherwise restore the tubular architecture. Capillary rarefaction, the principal driver of fibrosis, wherein diminished restorative ability of peritubular microvascular density, with a loss of 30-40%, driven partly by pericyte detachment and partly by sustained hypoxia, compounds this injury by creating an ischaemic microenvironment that further transitions into the ischaemic-reperfusion injury cycle. The fibrosis that results is not a remnant scar of a battle fought and recovered from. It is an archive of a malfunctioning repair program and of events ensuing from arrested epithelial cells converted to a phenotype promoting fibrosis (TGF upregulation). The tension here is not about whether kidney function dipped and regained, but a direct question about the fidelity of tubular epithelium repair-adaptively or arrest in a profibrotic state. Peak creatinine does not predict G2/M arrest, AKI staging does not predict capillary density at 90 days, and creatinine normalization does not comment on complete structural recovery or a dire state of reduced but functionally adequate nephron mass with ongoing subcortical fibrosis.
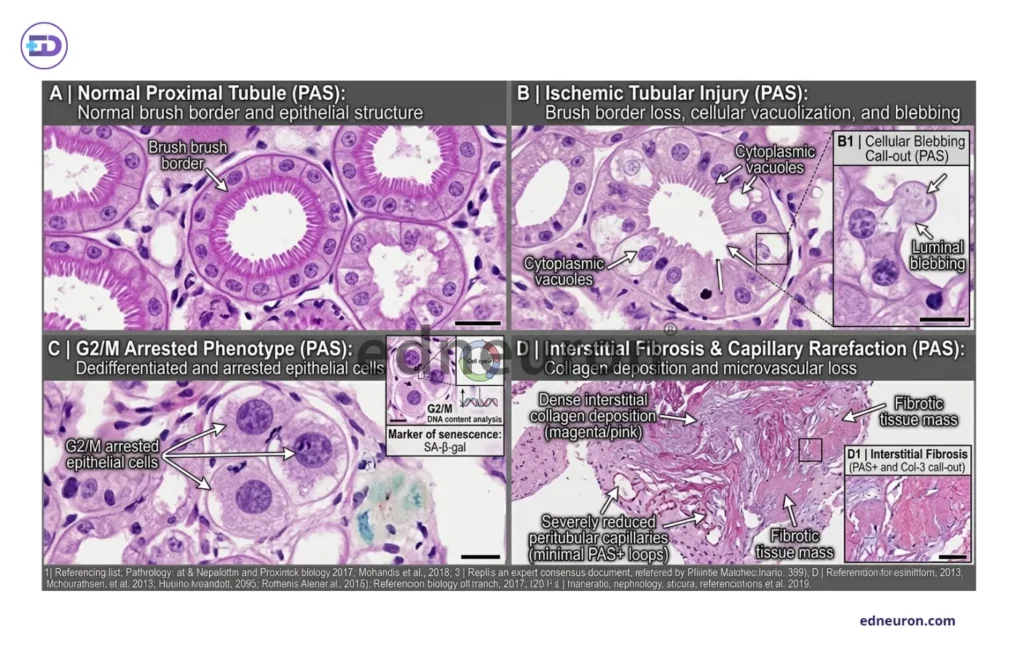
Fig 2 : A) Normal proximal tubular epithelium with preserved brush border morphology and intact epithelial polarity on PAS staining. B) Ischemic tubular injury characterized by brush-border loss, cytoplasmic vacuolization, epithelial simplification, and luminal blebbing, representing early structural manifestations of tubular damage. C) Tubular epithelial cells exhibiting features of maladaptive repair , including dedifferentiation and persistent G2/M cell-cycle arrest, associated with profibrotic signaling and impaired regenerative capacity. D) Chronic remodeling phase of interstitial collagen deposition, reduced peritubular capillary density, ECM expansion, and fibrosis.
Biomarkers: Promise and the Implementation Frontier
Tubular injury biomarkers have reshaped the conceptual language of AKI research without rewriting clinical practice at scale. KIM-1 is released on proximal tubular epithelial dedifferentiation and ischemic and toxic injury; NGAL, released from the distal injured nephron(neutrophil gelatinase-associated lipocalcin), rises within hours of tubular stress; and the TIMP-2/IGFBP7 product detects G1-phase cell-cycle arrest, a biologically distinct signal from frank necrosis or apoptosis, reflecting a tubular stress state that may precede maladaptive repair. These markers are not interchangeable because they represent different injury compartments, different timing windows, which vary more or less in a heterogeneous population. The current clinical task force provides no standardized values that can carry the diagnostic weight of the underlying pathology. Multiplex biomarker panels, combining markers of injury, outperform in cohort studies but remain immature because we still do not have assay standardization, decision thresholds are not universally validated, and the clinical action triggered by a high TIMP-2·IGFBP7 in the absence of a proven intervention remains ambiguous.
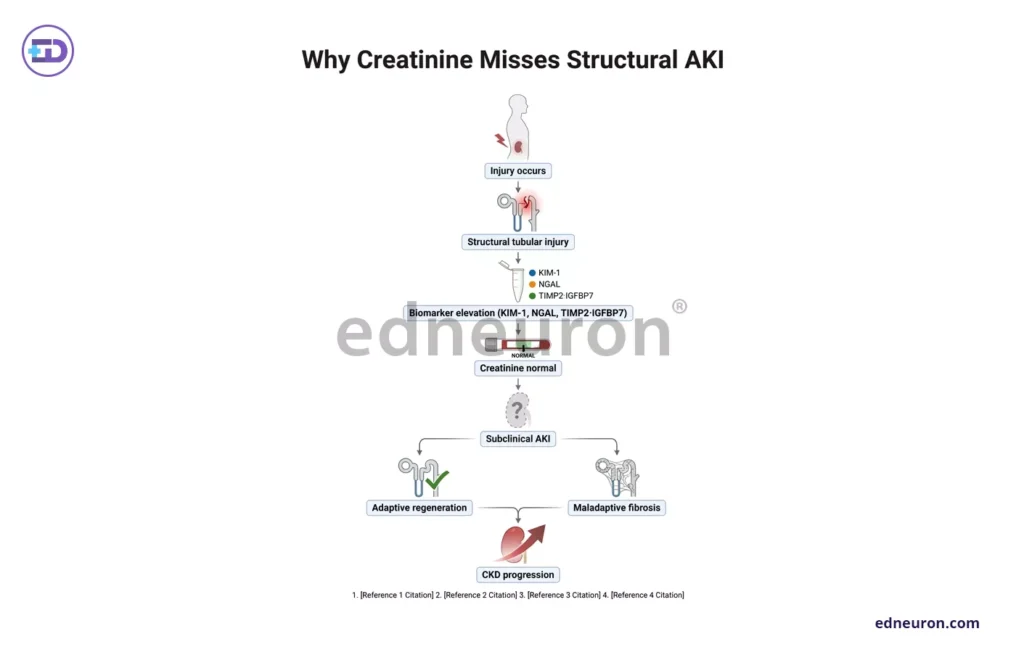
Fig 3 : Flowchart Represenation of why creatinine fails in subclinical AKI.
A Newer Classification for AKI Biology and Conclusion
The path forward is not to supplant creatinine staging and, in doing so, to refuse its indispensable simplicity and universal accessibility. It is to absolve any framework that advocates a unilayered categorization and to add dimensions to recognize a kidney that is molecularly plundered in its struggle for a normal routine creatinine signature. It is the demand of the era to integrate tools capable of detecting cortical fibrosis, such as multiplex biomarker profiles combined with urine microscopy and post-AKI eGFR.
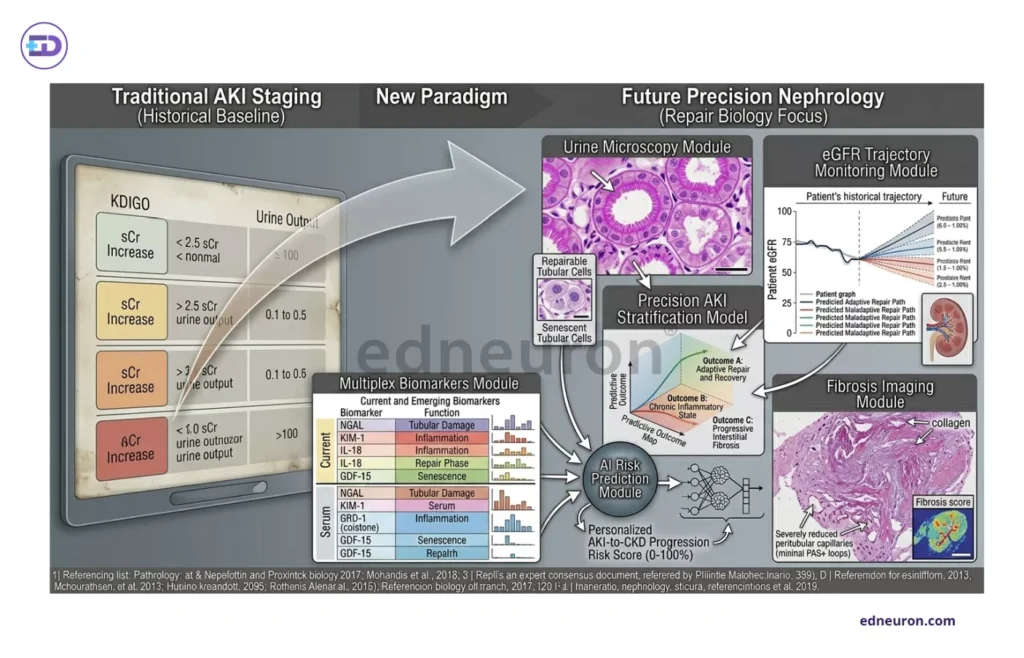
Fig 4 : The future of AKI risk stratification.
AKI stratification should be reimagined, where our capabilities and disposition should enable us to make accurate predictions and early management because the spectators of the next decade eye how nephrology maneuvers in the fast-moving realm of advanced diagnostics and innovative research; whether tubular epithelium chooses senescence over regeneration, and whether, at that decision point, clinical medicine was watching or not.
References
- KDIGO Clinical Practice Guideline for Acute Kidney Injury. Kidney Int Suppl. 2012;2(1):1–138. Available at: https://kdigo.org/guidelines/acute-kidney-injury/
- Chawla LS, Kimmel PL. Acute kidney injury and chronic kidney disease: an integrated clinical syndrome. Kidney Int. 2012;82(5):516–524. doi:10.1038/ki.2012.208
- Haase M, Devarajan P, Haase-Fielitz A, et al. The outcome of neutrophil gelatinase-associated lipocalin-positive subclinical acute kidney injury: a multicenter pooled analysis of prospective studies. J Am Coll Cardiol. 2011 Apr 26;57(17):1752-61. PMID: 21511111.
- Matarneh A, Akkari A, Sardar S, et al. Beyond creatinine: diagnostic accuracy of emerging biomarkers for AKI in the ICU – a systematic review. Ren Fail. 2025 Dec;47(1):2556295. PMID: 40983596.

Author: A P
A medical trainee with an emerging focus on translational and clinical research, with interests spanning surgical sciences, neuroscience, pediatrics, and immunology. Her academic trajectory reflects an effort to integrate molecular innovation with clinically relevant disease models, particularly in complex and high-burden conditions. Her research experience includes work in genome engineering, specifically in prime editing, exploring its therapeutic potential in precision medicine. She has also contributed to oncological research examining cholangiocarcinoma with brain metastasis, focusing on its clinical course and diagnostic challenges. In parallel, her work investigating stoma formation as an independent risk factor for acute kidney injury reflects an interest in perioperative and systemic complications. Academically, she has contributed to case-based and review-driven scholarship, including a case reports and interdisciplinary review articles. Her evolving interests in neurology, pediatrics, and immunology reflect a broader inclination toward understanding disease across systems—from molecular mechanisms to clinical outcomes—while maintaining a disciplined, evidence-based approach to patient care. MBBS (MS4) ABVIMS Dr. RML HOSPITAL New Delhi