
Abstract
VEXAS syndrome, the acronym for Vacuoles, E1 enzyme, X-linked, Autoinflammatory, Somatic, represents one of the most consequential nosologic revisions in modern medicine. Described first in 2020 in a cohort of 25 men by Beck DB et al. through a genotype, the first strategy using digital droplet polymerase chain reaction assay, immunoblotting, immunohistochemical testing, electron microscopy, flow cytometry, and cytokine profiling, this adult-onset hematoinflammatory disease collapses the traditional boundaries separating rheumatology, hematology, and immunology. The pathological engine is not an aberrant adaptive immune response but rather a somatically mutated hematopoietic clone harboring loss-of-function variants in UBA1, the master E1 ubiquitin-activating enzyme. Dysfunction in ubiquitin-mediated proteostasis that is lineage restricted, i.e., in myeloid and erythroid precursors, generates cytokine amplification, innate immune dysregulation, and a systemic inflammatory phenotype that convincingly masquerades as vasculitis, relapsing polychondritis, and myelodysplastic syndrome. This article means to ask a different question from most reviews: not what VEXAS is, but why medicine so consistently failed to recognize it.
Consider this Clinical Case
A man in his late sixties presents with auricular chondritis, febrile episodes, and a skin eruption resembling Sweet syndrome. His ESR and CRP are markedly elevated. Yet his antinuclear antibody and rheumatoid factor are negative. A bone marrow biopsy, performed reluctantly after his third hospital admission, reveals unexplained vacuolization of myeloid and erythroid precursors alongside macrocytic anemia and thrombocytopenia. He has been treated sequentially for relapsing polychondritis and seronegative vasculitis, although with failure. This patient did not have vasculitis. He did not have a primary rheumatologic disease. He had VEXAS syndrome, and the clues- older male, seronegative serology, macrocytic anemia, steroid dependence, marrow vacuoles, had been visible for years. The question is not why these clues were obscure. It is why medicine, organized as it is around organ systems and specialty guilds, was structurally incapable of reading them together.
Fig 1: Real time image illustration of patient with VEXAS syndrome.
How Come These Patients Have Remained Out of the System?
VEXAS syndrome was not something that didn’t exist. It was only misnamed systematically. When Beck and colleagues published their landmark 2020 discovery in the New England Journal of Medicine, they did so using a genotype-first strategy. They began by identifying UBA1 variants in an undiagnosed disease registry and working toward a phenotype that was not necessarily consistent across patients. The conventional phenotype-first approach, wherein clinical features generate a differential diagnosis that is then confirmed biologically, had failed these patients for decades. The trap here is multidimensional. VEXAS presents across four specialty boundaries simultaneously: rheumatology receives the auricular and nose chondritis with vasculitic rash; hematology has to deal with cytopenias and myeloid dyspoiesis; dermatology receives the neutrophilic dermatosis; and internal medicine receives the fever and elevated acute-phase reactants. No single specialty can pin a coherent answer until it deliberately seeks cross-domain synthesis. To add to the conundrum is the seductive partial response to glucocorticoids. VEXAS patients improve with steroids, fevers subside, skin lesions regress, CRP falls, producing exactly the reassurance that sustains a misdiagnosis. The flare that follows a steroid taper is attributed to disease recurrence rather than to re-examination, as evidence that the underlying mechanism has not been addressed. Steroid dependence, in the frame of rheumatology, is a management problem. Within the VEXAS framework, it is a diagnostic clue.
Bone Marrow: An Immunologic Organ
It is indeed not novel; we have well-documented accounts of pathologies in which cells exhibit somatic mutations and are then clonally selected, resulting in either benign hematologic conditions such as PNH or marrow failure syndromes. But mutations such as these occurring in men with a late-onset, treatment-refractory inflammatory syndrome with associated hematologic abnormalities continue to remain poorly understood in the context of autoinflammatory and rheumatologic diseases. UBA1, which encodes the primary E1 ubiquitin-activating enzyme, is the apex regulator of the ubiquitin-proteasome system. Ubiquitination not only governs protein turnover but also innate immune signaling, the DNA damage response, and the resolution of cellular stress. Loss-of-function somatic mutations at the methionine,41 codon, the dominant VEXAS lesion, eliminate or impair the cytoplasmic isoform of UBA1 (UBA1b), creating a state of profound proteostatic stress within hematopoietic progenitors. This defect was not found to be associated with aneuploidy on karyotype analysis, nor were X-chromosomal copy number variations present (determined by high-density single-nucleotide polymorphism array analysis). These were successfully predicted to be heterozygous somatic variants (also known as mosaic or postzygotic) mutations, with genetically heterogeneous cells carrying either hemizygous wild-type or mutated UBA1.
The consequences of such a mutation are layered. Misfolded and uncleared proteins accumulate, triggering unfolded protein response pathways and cytoplasmic vacuolization, the eponymous marrow finding that, when present, is nearly pathognomonic. The intolerance to haplosufficiency in UBA1 results in defective ubiquitylation that prevents the clearance of innate immune signaling debris: NF,κB activation persists; interferons and pro-inflammatory cytokines, including TNF,α, IL-6, and IL-1β, are amplified; and myeloid cells adopt a constitutively intrinsically activated inflammatory phenotype. Functional studies of macrophages and neutrophils showed preserved phagocytic capacity in mutant cells but enhanced spontaneous neutrophil extracellular trap formation, consistent with proinflammatory neutrophilic dermatoses and tissue infiltration that clinically resemble leukocytoclastic vasculitis, complete with perivascular infiltrates on biopsy, without the underlying vascular immunopathology of true vasculitis. Despite the mutation being lineage-restricted, the mosaicism arises de novo in early pluripotent hematopoietic progenitors in adult life. This is not germline autoinflammation; it is acquired clonal disease. The clone expands with age, displacing wild-type hematopoiesis and generating an increasingly dominant myeloid compartment hardwired for inflammatory output. The inflammatory phenotype, in this sense, is not occurring adjacent to the marrow clone; it is emerging from it. The bone marrow is not a bystander. It is the immunologic organ of disease.
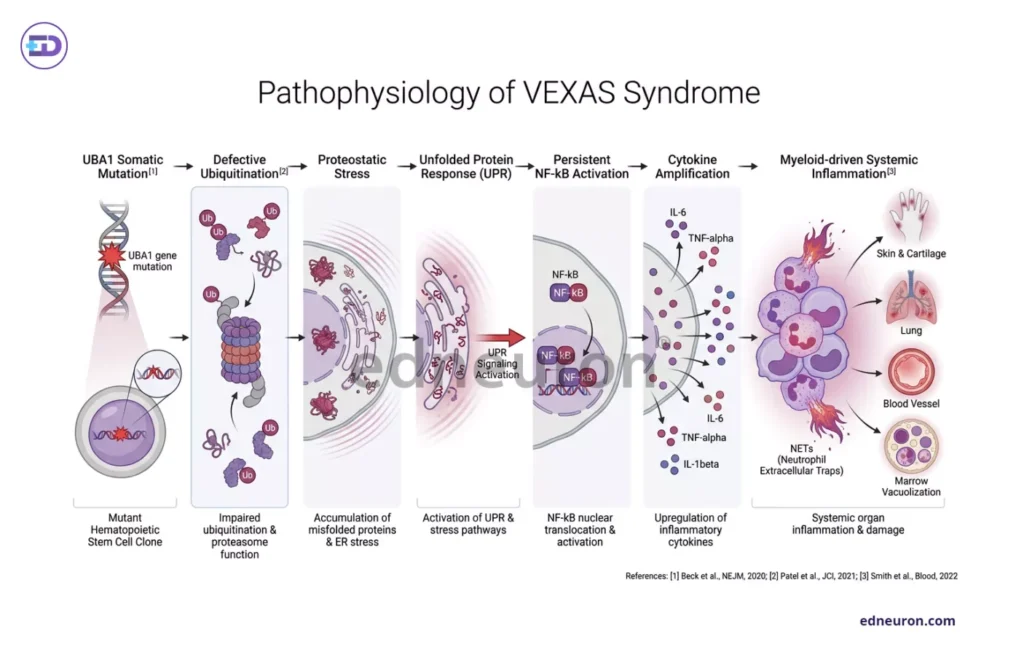
Fig 2: Stepwise flowchart illustrating pathophysiology of VEXAS syndrome.
The Liquid Interface of Hematology and Rheumatology
The blurry horizon of VEXAS and MDS is neither coincidental nor a mirage. Hematologists, encountering macrocytic anemia and marrow dyspoiesis, rarely ask whether the marrow itself could be the cause of systemic inflammation, even though a substantial proportion of VEXAS patients meet formal MDS criteria. MDS is understood as a disease of failed hematopoiesis, not of inflammatory excess, a frame that is mechanistically incomplete in light of VEXAS. Conversely, rheumatologists encountering a patient with auricular/nose chondritis, alveolitis, thromboembolic disease, and elevated inflammatory markers rarely find the marrow dubious. It is more commonly treated as a cartilage, and pulmonary,directed autoimmune disease with variable systemic features. The co-occurrence of macrocytosis and thrombocytopenia may be attributed to methotrexate toxicity or chronic inflammation rather than recognized as an independent hematologic disease. It is also undeniable that modern internal medicine is organized around the assumption that inflammation, when CRP and ESR are elevated in the absence of infection, is either autoimmune or autoinflammatory, and that hematologic abnormalities are downstream consequences rather than upstream drivers. VEXAS inverts this hierarchy, forcing the clinician to step outside its own specialty bubble and seek what lies beyond.
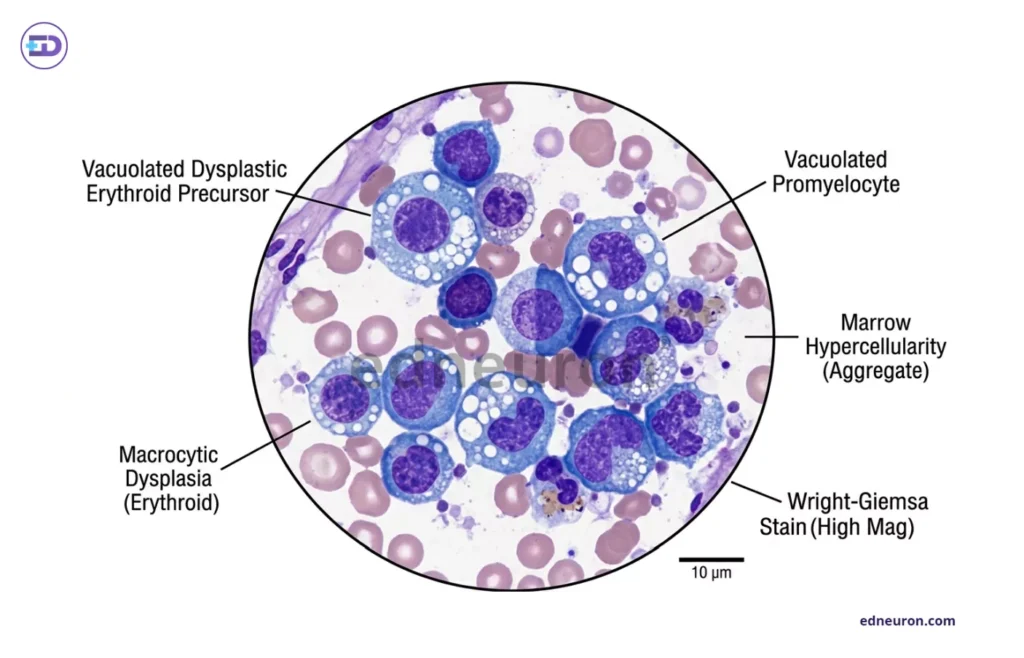
Fig 3: Hematopathology illustration of bone marrow aspirate in VEXAS syndrome showing vacuolization in myeloid and erythroid precursor cells, macrocytic dysplasia, marrow hypercellularity, and cytoplasmic vacuoles.
Therapeutic Limitations and Light of the Dawn
Standard immunosuppressive agents such as methotrexate, azathioprine, and mycophenolate act on differentiated immune cells and on lymphocyte-driven pathways. They do not address the mutant progenitor clone generating the inflammatory myeloid compartment. Data from the AIDA international registry document the limited efficacy of conventional DMARDs, even in combination with glucocorticoids, reporting high rates of treatment failure (31.8%) and the least favorable outcomes with TNF inhibitors. Targeted approaches that engage the inflammatory circuitry more directly- JAK inhibitors, IL-6 receptor blockade, and hypomethylating agents such as azacitidine have shown more promising signals. However, evidence remains largely retrospective. A conceptually more coherent approach would be to modify clonal dynamics to attenuate inflammatory output. Hematopoietic stem cell transplantation and genetic engineering using CRISPR perhaps remain the only treatments with the potential for durable disease modification. The 2026 landscape of VEXAS Therapeutics remains empirical and evolving. No randomized controlled trial has yet reported results. The disease, having been recognized for only a few years, is accumulating trial data that reflect its complexity, mandating enrolment of patients across hematology and rheumatology departments, stratification by UBA1 variant and MDS status, and the definition of endpoints that capture both inflammatory and hematologic disease burden.
A Reflection on Clinical Reasoning
VEXAS syndrome is, at its most vulnerable, an argument about how medicine develops its evaluation strategies. The disease existed before; what changed was the lens and the framework that allowed the data sets to be interpreted meaningfully. Thousands of patients carried VEXAS in their bone marrow while being treated for diseases they did not have, with therapies that could not find the source that plagued their minds and bodies. The research did not just hinge on the discovery of a new disease, but on the lesson it delineates: that discovery required abandoning the diagnostic frame that would have made it impossible. The bone marrow, once treated as a factory that supplied blood cells to the periphery, must be reconceptualized as an active immunologic organ, one whose somatic mutation landscape shapes the inflammatory tone throughout the body. VEXAS is the most vivid example of this reconceptualization, but it is unlikely to be the last. The precept is to keep eyes wide open and ask whether the inflammation originates in the marrow.
References
- Ahmed, I., Khan, M., & Rehman, A. (2024). Laparoscopic versus open appendectomy in complicated and uncomplicated appendicitis: A comparative study. Cureus, 16(3), e12433581.
- Beck DB, Ferrada MA, Sikora KA et al. Somatic Mutations in UBA1 and Severe Adult-Onset Autoinflammatory Disease. N Engl J Med. 2020 Dec 31;383(27):2628-2638. PMID: 33108101.
- Koster MJ, Warrington KJ. VEXAS within the spectrum of rheumatologic disease. Semin Hematol. 2021 Oct;58(4):218-225. PMID: 34802543.
- Steensma DP, Bejar R, Jaiswal S et al. Clonal hematopoiesis of indeterminate potential and its distinction from myelodysplastic syndromes. Blood. 2015 Jul 2;126(1):9-16. PMID: 25931582.
- Koster MJ, Warrington KJ. VEXAS within the spectrum of rheumatologic disease. Semin Hematol. 2021 Oct;58(4):218-225. PMID: 34802543.
- Fiumara M, Campochiaro C, Molteni R. Decoding VEXAS syndrome: emerging insights into pathogenesis and clinical management. Curr Opin Rheumatol. 2026 Jan 1;38(1):45-52. PMID: 41175032.

Author: A P
A medical trainee with an emerging focus on translational and clinical research, with interests spanning surgical sciences, neuroscience, pediatrics, and immunology. Her academic trajectory reflects an effort to integrate molecular innovation with clinically relevant disease models, particularly in complex and high-burden conditions. Her research experience includes work in genome engineering, specifically in prime editing, exploring its therapeutic potential in precision medicine. She has also contributed to oncological research examining cholangiocarcinoma with brain metastasis, focusing on its clinical course and diagnostic challenges. In parallel, her work investigating stoma formation as an independent risk factor for acute kidney injury reflects an interest in perioperative and systemic complications. Academically, she has contributed to case-based and review-driven scholarship, including a case reports and interdisciplinary review articles. Her evolving interests in neurology, pediatrics, and immunology reflect a broader inclination toward understanding disease across systems—from molecular mechanisms to clinical outcomes—while maintaining a disciplined, evidence-based approach to patient care. MBBS (MS4) ABVIMS Dr. RML HOSPITAL New Delhi